For over a century, alloy design followed one tradational rule: choose a single base metal and tweak its properties by diluting with small amounts of other elements.
A 2026 perspective paper published in Current Opinion in Solid State and Materials Science by Hickel et al. from the Federal Institute for Materials Research and Testing (BAM, Berlin) argues that this approach has reached its limits. Their work proposes a fundamentally different paradigm, built on chemical complexity rather than chemical dilution.
What Are Chemically Complex Materials and Why Do They Change Everything?
Chemically complex materials (CCMats) are defined by incorporating at least three principal elements, each present at more than 5 atomic percent. The most studied subset, high-entropy alloys (HEAs), typically contain five or more elements in near-equal proportions.
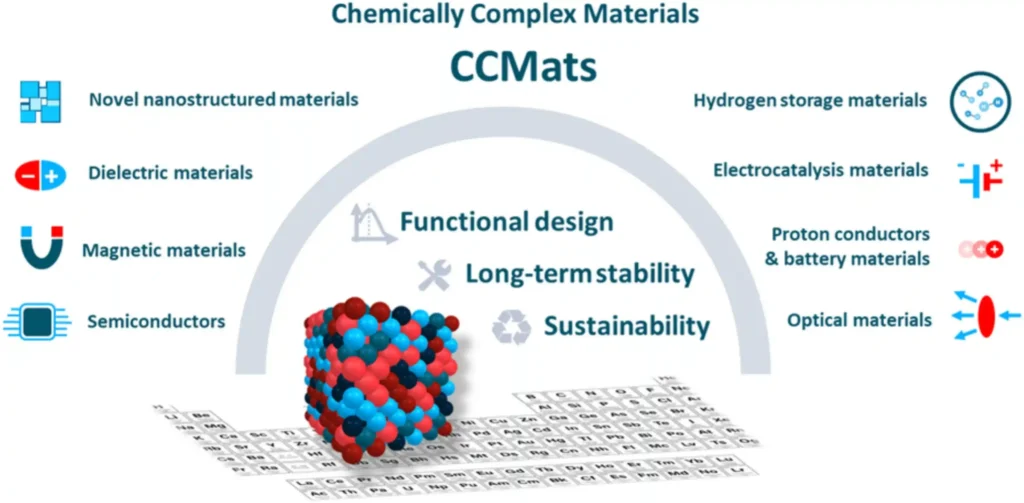
The result is a vast, largely unexplored compositional space with the potential to simultaneously optimize functional performance, long-term structural stability, and environmental sustainability.
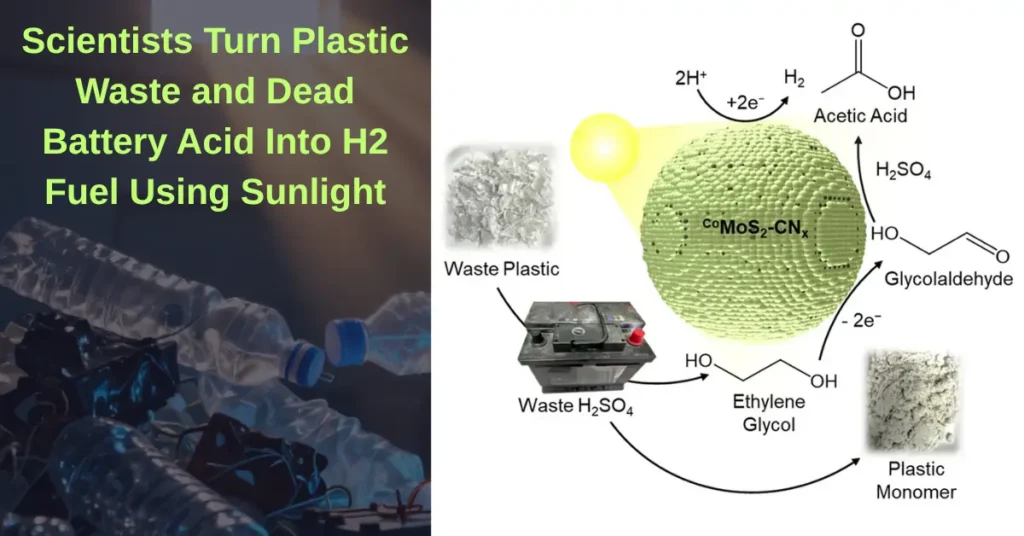
Related: Scientists Turn Plastic Waste and Dead Battery Acid Into H2 Fuel Using Sunlight
Why Traditional Alloy Design Falls Short
Single-phase HEAs were initially expected to dominate materials research due to their simple crystal structures and easy processability. Reality proved more complicated. Truly single-phase HEAs turned out to be extremely rare, and those that existed did not reliably outperform conventional alloys at room temperature.
The famous CrMnFeCoNi Cantor alloy, a benchmark five-component HEA, shows exceptional mechanical properties at cryogenic temperatures but underperforms at elevated temperatures. Solid solution strengthening alone, the mechanism that homogeneously distributes stress across a mixed lattice, cannot compete with precipitation hardening or grain boundary strengthening at room conditions.
The paper’s core argument is that research must shift beyond mechanical response. CCMats hold real value when their functional and structural performance are designed together, not in isolation.
Three Design Strategies That Drive CCMat Performance
The BAM team identifies three interlocking design strategies for CCMats:
- Diversity Management (DIV) targets compositional regions where good functional properties remain stable even under chemical fluctuations. This tolerance is especially important for recycled feedstocks, where exact compositions cannot always be controlled.
- Targeted Substitution (SUB) replaces critical, scarce, or toxic elements with more abundant alternatives. In permanent magnet research, for example, the team used ab initio thermodynamic calculations to screen all 3d and 4d metals as partial substitutes for neodymium in Ce-Fe-Ti based hard magnets. Zinc and titanium emerged as promising candidates for lowering the critical phase formation temperature from 710 K, making the material more thermally stable.
- Defect Engineering (DEF) treats microstructural features, including grain boundaries, dislocations, and stacking faults, as design variables rather than imperfections. In hydrogen storage, intentionally tuned defect densities can improve hydrogen uptake by generating additional interstitial sites under stress fields.
Related – Visit our thermodynamcis calculators
Related: Personal Carbon Footprint Calculator – Track your CO2 Emissions
The Science Behind Hydrogen Storage in CCMats
Hydrogen storage is one of the most demanding functional applications for CCMats. The key figure of merit is the hydrogen-to-metal ratio (H/M), representing how many hydrogen atoms a material can absorb per metal atom.
Formula:
H/M = N_H / N_M
where,
- H/M = hydrogen-to-metal ratio (dimensionless, higher is better for storage capacity)
- N_H = number of hydrogen atoms absorbed in the lattice
- N_M = number of metal atoms in the alloy
In plain words: H/M quantifies storage density at the atomic level. A value of 1.0 means one hydrogen atom per metal atom is absorbed, while theoretical maximums in bcc structures can exceed this significantly.
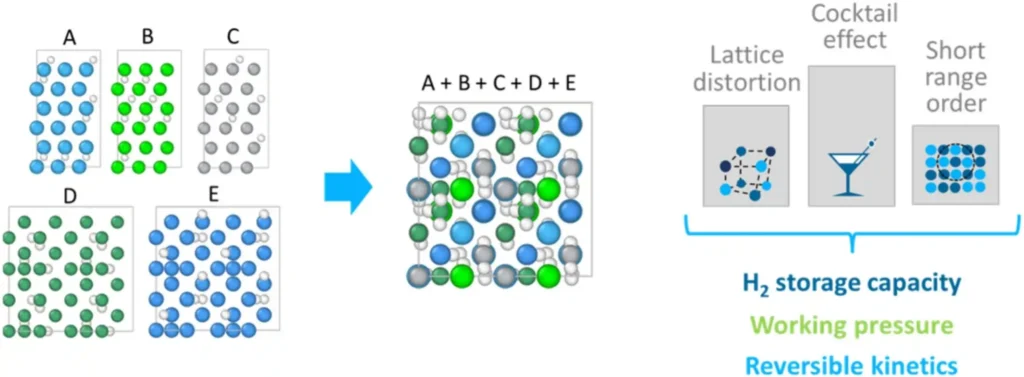
Two physical mechanisms drive high H/M values in CCMats. First, lattice distortion from size-mismatched elements creates local interstitial volumes that can trap hydrogen. Second, the “cocktail effect,” the collective interaction of multiple elements in one lattice, generates deep energetic traps that hold hydrogen more securely.
Chen et al. demonstrated this with the TiZrFeMnCrV C14-phase alloy, which maintained H/M = 1.05 over 50 charge-discharge cycles without significant degradation.
Key Performance Data Across Application Areas
The research covers eight distinct application domains. Selected representative results are shown below.
| Application | Material | Key Result | Significance |
|---|---|---|---|
| Hydrogen Storage | TiZrFeMnCrV (C14 phase) | H/M = 1.05 over 50 cycles | Stable cyclic capacity without degradation |
| Li-ion Battery Cathode | Li1.3Mn0.1Co0.1Cr0.1Ti0.1Nb0.2O1.7F0.3 | 307 mAh/g at 20 mA/g | Matches commercial cathodes with lower cobalt |
| Proton Conductor | BaSn0.16Zr0.24Ce0.35Y0.1Yb0.1Dy0.05O3-delta | 8.3 x 10^-3 S/cm at 600 °C | High conductivity in intermediate temperature range |
| Dielectric Energy Storage | KNN-H high-entropy ceramic | Wrec = 10.06 J/cm3, eta = 90.8% | Exceeds conventional lead-free ceramics |
| Thermoelectric | AgSnSbSe1.5Te1.5 | Average ZT = 1.0 over 400-773 K | Broad temperature range with no phase transition |
| Oxide Glass (CCMat) | R2O3-Y2O3-TiO2-ZrO2-Al2O3 | Hardness = 12.58 GPa | Highest reported microhardness for an oxide glass |
Also Read: The Crucial Role of Chemical Engineering in Everyday Life
Also Read: Online Psychrometric Calculator for Chemical Engineers
Computational Tools Accelerating CCMat Discovery
Navigating a compositional space with five or more elements requires computational methods that go far beyond traditional trial-and-error. The BAM team highlights three tiers of simulation tools used in their workflow.
- Density Functional Theory (DFT) calculates ground-state energies and phase stabilities from quantum mechanics. For CCMats, DFT uses supercell representations called Special Quasirandom Structures (SQS) to simulate random atomic mixing. The limitation is computational cost: DFT scales poorly beyond roughly 100 atoms per simulation cell.
- Machine Learned Interatomic Potentials (MLIPs), trained on DFT data, extend atomistic simulations to millions of atoms at a fraction of the cost. Foundation models like MACE-MP-0 and CHGNet, pre-trained on large materials databases, can predict free energies and functional properties across much of the periodic table. This capability directly supports the SUB and DIV design strategies by screening thousands of compositions rapidly.
- CALPHAD (Calculation of Phase Diagrams) integrates thermodynamic databases for multi-component systems to map phase stability across compositions and temperatures. The team demonstrates CALPHAD combined with elastic energy corrections for the FeMnNiCoCu system, identifying spinodal decomposition regions at 723 K that convert an antiferromagnetic Cantor alloy variant into a magnetically functional material by substituting Cr with Cu. Explore thermodynamic modeling tools relevant to phase equilibria on chemenggcalc.com.
Related Article: How AI and Self-Driving Labs are Transforming Chemical Engineering
What Stands Between CCMats and Industrial Use
Several barriers remain before CCMats reach broad deployment. Characterizing chemical short-range order (SRO), the tendency of certain element pairs to cluster or avoid each other at the atomic scale, requires multimodal techniques. No single probe resolves SRO unambiguously in a five-component system.
X-ray absorption spectroscopy (XAS) gives element-selective pair distances, while neutron pair distribution function (nPDF) analysis provides complementary contrast for light elements. Combining both with Reverse Monte Carlo simulation currently represents the most reliable structural analysis route.
Thin-film CCMats for coatings and electronics present an additional challenge: existing reference materials and measurement protocols designed for simple alloys do not transfer to multi-element films. BAM is actively developing Cantor alloy thin-film reference materials with thicknesses from 10 to 500 nm to address this gap.
Scalable synthesis is a third constraint. High-entropy semiconductors, for instance, typically require synthesis temperatures above 1000 °C. Recent work by Folgueras et al. demonstrated room-temperature solution synthesis of cubic Cs2MCl6 high-entropy halide perovskite single crystals, offering a potential path toward lower-energy processing routes.
The Road Ahead for Chemically Complex Materials
The perspective from Hickel and colleagues at BAM makes a clear case: the materials science community must integrate high-throughput experiments, machine learning, multimodal characterization, and CALPHAD-based thermodynamics into unified design workflows.
Read more about recent chemical engineering materials breakthroughs on the chemenggcalc research blog.
CCMats are not a single material class but a design philosophy. By choosing compositions that are tolerant to chemical fluctuations, substituting critical elements systematically, and treating defects as functional assets, engineers can access property combinations that dilute alloys simply cannot reach.
The next generation of wind turbine magnets, solid-state battery electrolytes, and radiation-resistant optical glass may all trace their origins to this expanding field.
For engineers working on composition screening and materials property calculations, the methods described in this paper offer a structured framework applicable across virtually every functional material category.
Original Research: Chemically complex materials enable sustainable high-performance materials — Hickel, T., Waske, A., Tehranchi, A., et al. Current Opinion in Solid State and Materials Science, 42 (2026) 101256. Open access under CC BY 4.0.